Uncovering transcriptional and co-transcriptional regulatory programs in normal and diseased states
1) Impact of non-coding variants of protein-coding genes on cellular functions – Conventionally, transcription of RNAs from genomic loci of protein-coding genes are widely accepted to be translated into proteins. However, our recent work revealed that a large number of protein-coding genes generate RNA variants that retain little or none of the coding region, and hence represent a class of non-coding transcripts. We established that the expression of these non-coding RNA (ncRNA) variants is robust, regulated across normal cell types and dysregulated in cancer affirming that they do not represent ‘cryptic’ events or ‘transcriptional noise’. With the discovery of ncRNAs of protein-coding genes (ncp-RNAs) and initial evidence of their interactions with RNA binding proteins, we are investigating the functional role of ncpRNAs in diverse cellular processes at the transcriptional or post- transcriptional level by cis– or trans-regulation of gene expression programs. We are combining pooled perturbation approaches with next-generation sequencing to dissect the functional roles of these ncpRNAs.
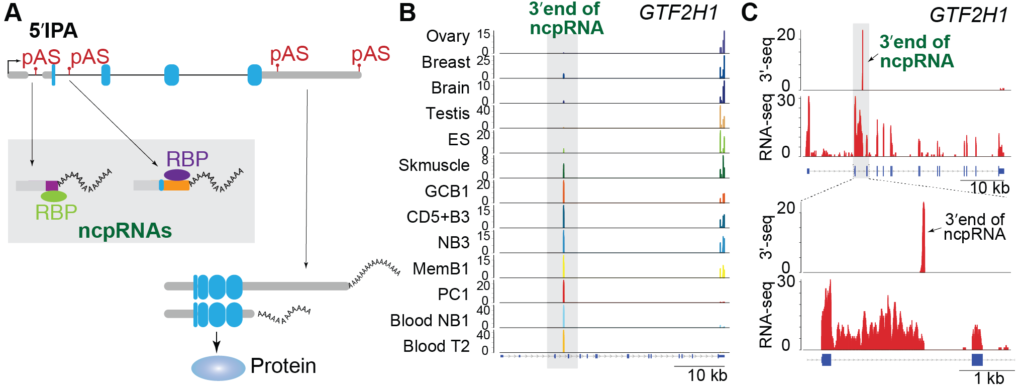
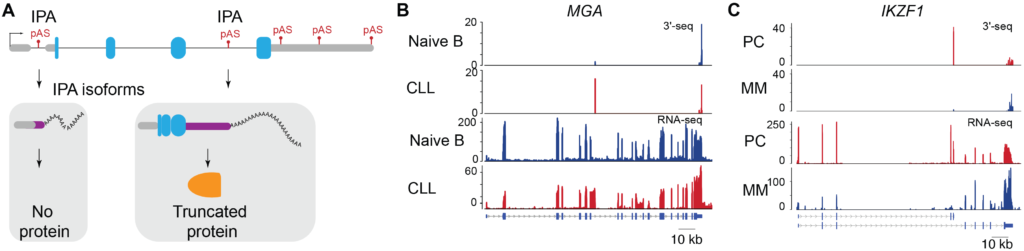
2) Dysregulation of RNA Processing as Driver of Malignancies – Dysregulation of intronic polyadenylation (IPA) is emerging as a novel pathobiological phenomenon in cancer. Recognition of polyadenylation signals (pAS) present in introns of protein-coding genes can generate truncated mRNAs (IPA isoforms) that are either non-coding variants or transcripts with truncated open reading frames that lead to loss of C-terminal domains in the protein product. Our recent studies showed that expression of IPA isoforms is dysregulated in Chronic Lymphocytic Leukemia (CLL) (Lee* and Singh* et al, Nature 2018) and Multiple Myeloma (MM) patients (Singh et al, Nature Communications 2018). We showed that truncated mRNAs generated by IPA is a widespread phenomenon in CLL patients and predominantly inactivates tumor-suppressor genes (TSGs). Inactivation of TSGs by aberrant mRNA processing was more prevalent than the loss of such genes through genetic events. In contrast to CLL, MM patients displayed a striking loss of IPA isoforms that were expressed in plasma cells (PC, normal cell type for MM). We discovered that dysregulated IPA expression in MM patients is associated with shorter progression-free survival. Interestingly, IPA dysregulation impacted key genes of MM biology that are involved in response to lenalidomide therapy (a highly successful MM therapeutic). Still, the functional consequence of this loss of IPA isoform expression in MM remains unknown and requires in-depth investigation. Overall our studies highlight that mRNA events can be widespread contributors to cancer pathogenesis. Thus, it is critical to identify target genes subject to IPA dysregulation across malignancies and determine their role in tumorigenesis. To accomplish this, our lab is interested in characterizing the landscape of IPA across malignancies and interrogating its functional consequences.
Dissect epigenetic and transcriptional regulatory programs
1) Computational modelling of chromatin directed gene expression profiles – Accumulation of genetic and epigenetic alternations leads to widespread changes in the gene expression programs in cancer. Aberrant activity of transcription factors (TFs) is instrumental in driving such gene expression changes by altering the chromatin accessibility landscape resulting in acquisition of hallmark capabilities of cancer: sustained proliferation, replicative immortality and apoptotic evasion. Data-driven computational methodlogies integrating the DNA sequence accessibility across promoters, intronic and intergenic enhancers can be effective to explain the gene expression profiles. We utilize learning framework to identify TFs that explain the gene expression either in a gene- or patient- specific manner.
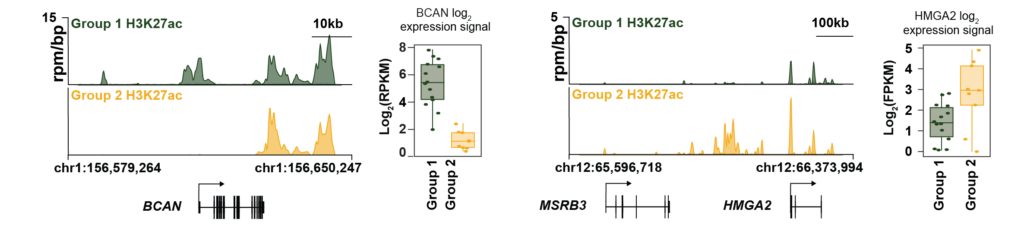
2) Understanding chromatin directed oncogenic transformation – Cancers are fundamentally believed to be caused by genetic alternations. However, recent studies have shown that changes in the epigenome have a strong potential to drive the fate of normal cells towards tumorigenic state. Specifically, changes in the epigenome can lead to acquisition of large clusters of active enhancers, also referred as “super-enhancers”. Super-enhancers are in the proximity of oncogenes resulting in multi-fold increase of their expression levels. Many of these super-enhancers drive expression of transcription factors (TFs) which in turn can directly regulate the expression of other oncogenes. In certain cancers and especially brain tumors, efforts to identify and target driver mutations have produced mixed results. Glioblastoma multiforme (GBM) lacks actionable targets owing to its high genetic and phenotypic heterogeneity. This calls for alternative strategies to discover new druggable targets predominantly in a non- genetic context. Our current efforts are focused on probing epigenetic profiles to identify non-mutated tumor dependencies, and specifically, super-enhancer associated drivers in glioblastoma. Our preliminary data shows the existence of two distinct super-enhancer states in a large series of Glioblastoma Stem Cell (GSC) models. We also observe de novo super-enhancers in GSCs compared to the normal neural stem cells (NSC). These data suggest that super-enhancer acquisition may be a key feature of GBM and motivates us to systematically characterize the oncogenic role of candidate TFs as drivers of tumorigenesis in glioblastoma and identify their downstream target oncogenes and pathways as candidate therapeutic targets.
© 2020 by The Singh Laboratory @ Texas A&M University. All rights reserved.